
Atoms, the fundamental building blocks of matter, are intricate quantum systems with a positively charged nucleus encircled by negatively charged electrons. When atoms unite to form molecules, their interactions become incredibly complex and difficult to simulate. For researchers, this mathematical labyrinth can sometimes seem insurmountable. Traditional computational methods, reliant on the Schrödinger equation—a cornerstone of quantum mechanics—have been the go-to approach for modeling these systems. However, even for relatively simple molecules, these calculations can spiral into an exhaustive endeavor, consuming vast amounts of computational power and time.
The challenges aren’t just technical; they pose a philosophical dilemma about our understanding of the very nature of matter. If we wish to delve deeper into the molecular realm, advancing materials for solar panels or designing new drugs, we must enhance our methods for simulating these essential processes. Yet, overcoming the computational bottleneck of molecular dynamics simulations has been a long-studied problem in science.
A Breakthrough Approach: Machine Learning Meets Molecular Dynamics
In an inspiring collaboration between the Berlin Institute for the Foundations of Learning and Data (BIFOLD) and Google DeepMind, a groundbreaking machine learning algorithm has emerged that would dramatically redefine molecular dynamics simulations. This new model promises increased accuracy while drastically improving computational efficiency, making long-term simulations accessible to researchers worldwide.
The traditional route of solving the Schrödinger equation isn’t merely challenging; it becomes nearly impractical when simulating interactions involving hundreds or thousands of atoms. Researchers have often found themselves at a crossroads, faced with the insurmountable task of running these complex calculations over vast timescales. Recent advancements in machine learning now provide a compelling alternative. Instead of wrestling with the Schrödinger equation directly, the new approach learns to predict electronic interactions based on previous data—essentially shortcutting the extensive computational requirements.
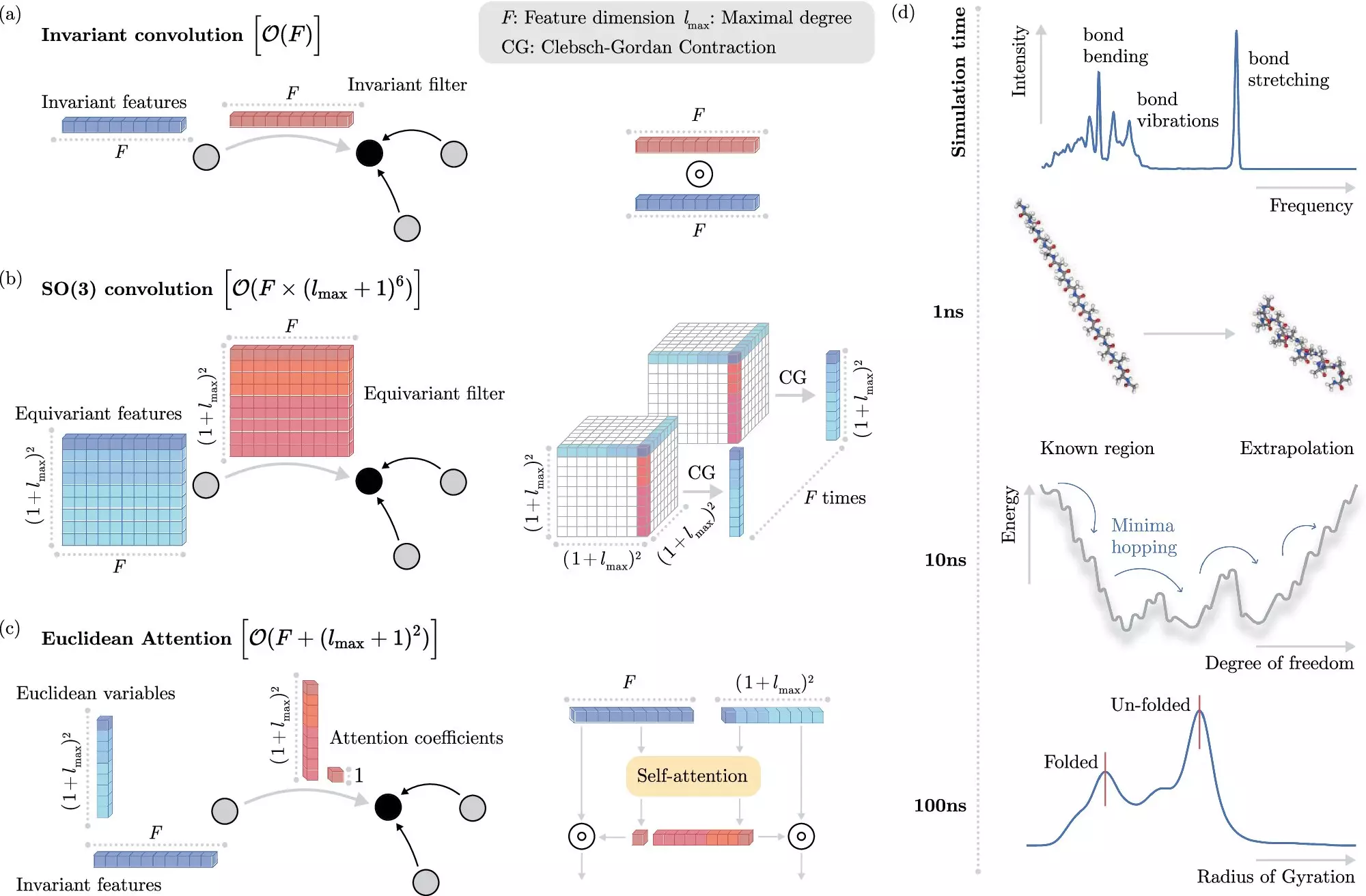
Unlocking Invariance: A New Paradigm
One of the pivotal aspects of the BIFOLD and Google DeepMind research lies in how the algorithm deals with invariances. In layman’s terms, invariance suggests certain physical traits of a molecule remain unchanged as it moves, provided the distances between its constituent atoms remain constant. Harnessing this principle intelligently allows researchers to streamline the required computations.
Previously, machine learning models incorporated these invariances in a manner that was still computationally burdensome. However, the team developed a method to decouple invariances from other chemical system information early in the process. This shift optimizes the computational task, allowing machines to manage the intricate operations of important physical phenomena without losing the vital details that matter.
Dr. Stefan Chmiela, a lead researcher on the project, eloquently states that the newfound efficiency can take computations that would traditionally last months or years and compress them into mere days on a single computer node. This capacity to simulate over extensive timescales could open gateways to understanding the very foundation of molecules, reshaping how we study complex systems in both chemistry and biology.
Practical Implications: From Drug Design to Material Science
Envision a future where researchers can test molecular interactions within the human body without the need for time-consuming and often expensive laboratory experiments. This revolutionary algorithm can help realize just that. The potential to simulate how drugs interact with proteins opens avenues for quicker development cycles and more environmentally friendly research practices—yielding effective treatments without the traditional trial-and-error processes.
To demonstrate the computational prowess of the new machine learning method, researchers focused on docosahexaenoic acid (DHA), an essential fatty acid vital for brain structure. Previously, determining the molecule’s most stable version required scanning extensive databases of potential candidates—a task traditionally beyond the reach of conventional quantum mechanical methodologies. The advancements from the BIFOLD team showcase the algorithm’s ability to navigate these complexities with superior precision.
The Road Ahead: Challenges and Future Directions
While this breakthrough undoubtedly marks a significant stride forward, it also prompts deeper inquiries into the next generation of molecular simulations. The implications extend beyond mere efficiency; they beckon a re-evaluation of how we understand complex systems at a molecular level. The pursuit of additional refining in the description of intricate long-range interactions will demand continuous innovation in both algorithm development and computational methodologies.
As we stand on the cusp of breakthroughs in computational chemistry, the combination of advanced machine learning techniques with physical principles serves not only as a tool for researchers but as an invitation for new paradigms in scientific understanding. The inherent beauty of this work lies in its potential to bridge gaps in knowledge and usher in a renaissance in molecular dynamics that could lead to unprecedented discoveries in fields yet to be explored. The road ahead may be challenging, but the prospects are luminous, signaling that we are closer than ever to unraveling the sophisticated dance of atoms and molecules that form the very fabric of our universe.